Autophagy is a cellular cytoplasmic degradation mechanism in which cell recycles broken or unnecessary proteins and organelles in the cell to get rid of these products. In order to save homeostasis and survival throughout times of metabolic stress, these intracellular essentials are recycled and provide an additional energy source. Autophagy allows longer survival in tumor cells that have apoptotic aberrations. Paradoxically, abnormal autophagy is linked to a higher risk of tumor development, albeit the exact mechanism is still unknown (Mathew et al., 2007). Although in some situations autophagy destroys tumorigenesis, in most contexts autophagy enables tumorigenesis. Cancers can upregulate autophagy to survive microenvironmental stress and to increase growth and violence. Mechanisms by which autophagy helps cancer include defeating induction of the p53 tumor suppressor protein and keeping metabolic function of mitochondria. Efforts to inhibit autophagy to recover cancer therapy have thus involved great interest (White, 2015).
Among the several autophagy-related effectors that perform the firmly controlled process of autophagy are ATG proteins. The first committed phase in autophagy, known as vesicle nucleation, is surrounding selected macromolecular meetings for devastation with distinct membranes called phagophores. Vesicle nucleation is carried out by the class III phosphatidylinositol-3-kinase (PI3Kc3 or VPS34), which stimulate the conversion of phosphatidylinositol to phosphatidylinositol 3-phosphate; the PI3Kc3 controlling subunit (p150 or VPS15), a myristylated serine/threonine kinase that phosphorylates PI3Kc3 and attractions it to the membrane; and the BCL-2 interacting protein (Beclin 1 or ATG6), which appears to be a center for protein interactions. Additional recently, it has been confirmed that Ambra 1, a progressive autophagy regulator that works with Beclin 1, is further to the core complex (Maria Fimia et al., 2007). Furthermore, by connecting in different ways with the vital complex, many other proteins like ATG14, UV radiation resistance-associated gene (UVRAG), vacuole membrane protein 1 (Vmp1), endophilin B1 (Bif-1), and Beclin 1 related RUN domain comprising protein (Rubicon) form middles with separate roles in membrane trafficking processes (He & Levine, 2010). Multidimensional vesicles called autophagosomes are formed when the membranes of budding phagophores formed and then fuse at their boundaries. This way involves two different complex conjugation mechanisms that resemble ubiquitin (Maiuri et al., 2007). The covalent joining between the E2-like enzyme ATG10 and the E1-like enzyme ATG7 in a specific formal. Then, ATG16 and the ATG12-ATG5 conjugate combine to form a larger multimeric composite. In the different system, three enzymes—an E1-like, ATG7; an E2-like; and a protease, ATG4—work in series to conjugate phosphatidylethanolamine to another ubiquitin-like protein, ATG8/LC3. When the external membrane of autophagosomes unites with lysosomes, autophagolysosomes are finally produced. Particular ATG8 complex, a number of ATG18 with WD repeat domains that cooperate with the phosphoinositides, ATG2A, a protein that work together with ATG18, ATG9, a transmembrane protein, SNX18, which improvements autophagosome tubulation, and small G-proteins that control this process are all involved in this phase. The autophagosome’s fillings are broken down by lysosomal enzymes inside the autophagolysosome. The cell then recycles the breakdown products (Su et al., 2013). The development of autophagy is the removal of biological material to lysosomes for breakdown, which outcomes in the basal turnover of cell elements and provides energy and precursors for macromolecules. There have been offers for cancer medicines that purpose to activate or inhibit autophagy, as autophagy plays inconsistent functions in cancer that vary depending on the situation. As a result, some scientists reflect that therapeutically targeting autophagy in cancer is doubtful (Levy et al., 2017). According to He and Klionsky (2009), macroautophagy enables a cell’s ability to respond to a diversity of extracellular and intracellular challenges, such as lack of nutrients, the presence or absence of insulin and other growth hormones, hypoxia, and ER stress.
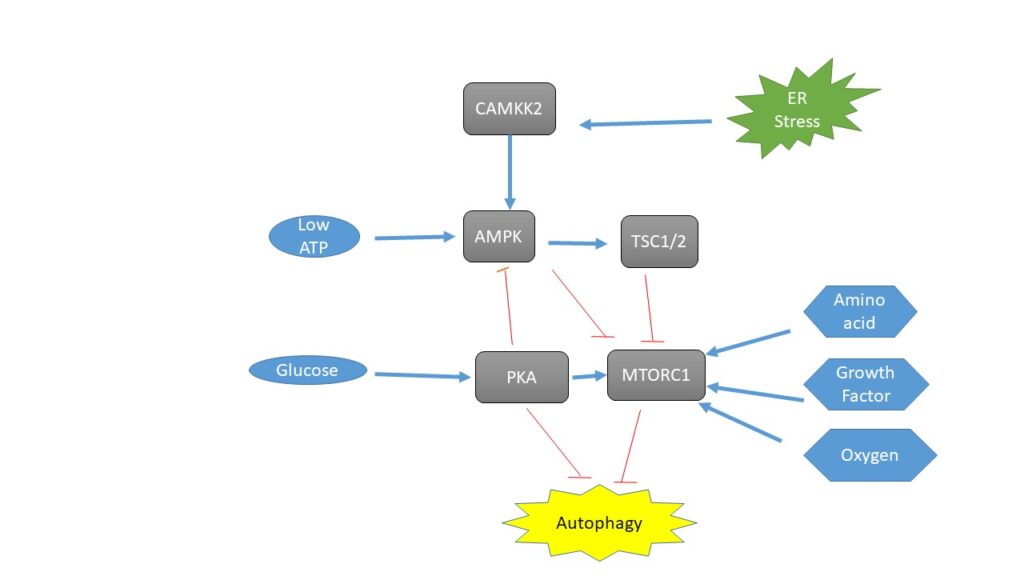
The cAMP-dependent protein kinase A (PKA) and mTOR pathways, which intellect principally carbon and nitrogen, respectively, control two pathways concerned in nutritional shortage. PKA inhibits macroautophagy in yeast when the culture is nutrient-rich. Mammals involvement this inhibition, at least in part, as a result of PKA phosphorylating LC3. Amino acid concentration positively regulates MTORC1 for its function in nitrogen sensing. RAG proteins, which are RAS-related small GTPases that activate MTORC1, are controlled by amino acids concentration (Parzych & Klionsky, 2014). Through MTORC1, growth factor concentrations and hypoxia both influence macroautophagy, and hypoxia can hinder MTORC1 even in the presence of enough growth factors and nutrition (2, 4). The intricate control of macroautophagy by several cellular signaling pathways makes MTORC1’s role in this process a very captivating and active field of study. This topic is enclosed in more detail in another review in this Medium series (Parzych & Klionsky, 2014). Additional marks such as hypoxia and the lack of growth hormones activate the onset of macroautophagy. The outline of macroautophagy occurs when growth hormones are absent, even in the presence of enough nutrition (Lee et al., 2011).
Autophagy is primarily regulated by PI3K/AKT/mTOR, Beclin-1, p53, and JAK/STAT signaling pathways. These autophagy-related controlling pathways have a theatrical impact on EMT. In the process of EMT, there are numerous signaling pathways, including integrin, WNTs, NF-kB, and TGF-β signaling pathways, that play a vital role in autophagy. Also, growing observations have designated that the functional contact between cytoskeleton and mitochondria is also the critical regulatory mechanism in the process of autophagy and EMT (Chen et al., 2019)

